| wzór strukturalny | |||||||
|---|---|---|---|---|---|---|---|
| K | |||||||
| Ogólne | |||||||
| Nazwa | Azotan potasu | ||||||
| Inne nazwy |
| ||||||
| Formuła molekularna | KNO3 | ||||||
| Numer CAS | 7757-79-1 | ||||||
| Krótki opis | biały krystaliczny proszek | ||||||
| Właściwości | |||||||
| Masa molowa | 101,11 g mol | ||||||
| Stan materii | naprawiono | ||||||
| Gęstość | 2,109 g cm (16°C) | ||||||
| Temperatura topnienia | 334 °C | ||||||
| Temperatura wrzenia | Rozkład>400 °C | ||||||
| Rozpuszczalność | dobra w wodzie (316 g/l w 20 °C), słaba w rozpuszczalnikach niepolarnych | ||||||
| Instrukcje bezpieczeństwa | |||||||
| |||||||
| Tam, gdzie jest to możliwe i powszechne, używane są jednostki SI. O ile nie zaznaczono inaczej, podane dane dotyczą standardowych warunków. | |||||||

Azotan potasu , lepiej znany jako Saltpeter w mowie potocznej , konkretnie jako azotan potasu , jest solą potasową kwasu azotowego.
Właściwości
Azotan potasu tworzy bezbarwne kryształy, które przy silnym chłodzeniu rozpuszczają się w wodzie. Dlatego jest znacznie łatwiej rozpuszczalny w ciepłej wodzie niż w zimnej. Do 130g azotanu potasu można rozpuścić w 1 litrze wody o temperaturze 0°C i do 2455g azotanu potasu w 1 litrze wody o temperaturze 100°C. Przy tak wysokich stężeniach gęstość roztworu jest znacznie większa niż w przypadku czystej wody.
Azotan potasu rozkłada się na azotyn potasu i tlen podczas ogrzewania:
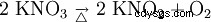
Jest doskonałym środkiem utleniającym w podwyższonych temperaturach. Zwęglone pozostałości w naczyniach szklanych szybko rozpuszczają się w stopionym azotanie potasu.
Azotan potasu jest znacznie mniej higroskopijny niż wiele innych azotanów, m.in. B. Azotan sodu.
Ekstrakcja
- Zdarzenia naturalne
Azotan potasu (min. „Nitrokalit”) występuje na glebach w postaci wykwitów. Znaczenie gospodarcze miały złoża w Chinach i Azji Południowo-Wschodniej, gdzie w pierwszej połowie XIX wieku wydobywano ponad 10 000 ton saletry rocznie poprzez ługowanie takich gleb.
- Nitryfikacja bakteryjna bogatych w azot odpadów organicznych Od końca XIV do XIX wieku saletra była produkowana w Europie przy pomocy bakterii i tlenu atmosferycznego, aby uniezależnić się od importu tego niezbędnego do celów wojennych surowca. (patrz „Historia”). Odpady organiczne bogate w azot (odchody i mocz) miesza się z wapnem i popiołem drzewnym (potażem) i pozostawia do rozkładu w luźnych, przepuszczalnych dla powietrza hałdach ziemi. Związki azotu są przekształcane przez bakterie w azotany. Po dwóch latach masa jest wypłukiwana wodą. Potaż jest dodawany do surowego ługu, przekształcając w ten sposób azotan wapnia i magnezu w azotan potasu i słabo rozpuszczalny węglan metali ziem alkalicznych. Azotan potasu otrzymuje się przez odparowanie przefiltrowanego ługu, który jest następnie oczyszczany przez rekrystalizację.
- Saletra konwersji
Od połowy XIX wieku do około 1920 roku konwersja chilijskiego azotanu w chlorek potasu była najważniejszym procesem produkcji azotanu potasu:NaNO3 + KCl --> KNO3 + NaCl
Wykorzystano tu nieznaczny wzrost rozpuszczalności chlorku sodu wraz z temperaturą:ług macierzysty KNO3 Krystalizacja w poprzednim cyklu jest podgrzewana i dodawana z surowym azotanem sodu i chlorkiem potasu w stosunku stechiometrycznym. Mieszaninę zatęża się, dodając niewielką ilość sody w 100°C, przy czym chlorek sodu i zanieczyszczenia (węglany metali ziem alkalicznych) wytrącają się i są odfiltrowywane. Przesącz ponownie rozcieńcza się kondensatem z odparowania w celu uniknięcia wytrącania soli sodowych podczas chłodzenia, odsącza przezroczystość, a następnie schładza do 5°C w celu skrystalizowania azotanu potasu i odwirowuje. Oddzielony azotan potasu poddaje się rekrystalizacji do celów technicznych.
- Syntetyczny z kwasu azotowego
Obecnie azotan potasu jest produkowany syntetycznie (patrz Przedstawiciel ).
Wygląd (tworzenie)
Istnieje wiele sposobów przedstawiania azotanu potasu:
- przez prawie wszystkie reakcje tworzenia soli:
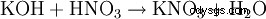

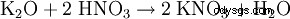
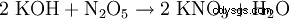
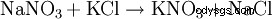
- z węglanu potasu:
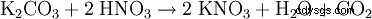
Wykorzystanie
- Azotan potasu służy do konserwowania żywności (sól peklująca E 252)
- Służy do produkcji czarnego prochu i innych mieszanek pirotechnicznych
- Mieszanka 24% boru + 71% KNO3 + 5% spoiwo (PMMA) służy jako niezawodna mieszanka do zapłonu, która pali się również w bardzo niskich temperaturach (-196°C).
- Mieszanka 60% NaNO3 +40% KNO3 topi się w 222°C i jest stosowany jako nośnik ciepła w elektrowniach słonecznych. Ta stopiona sól jest chemicznie stabilna do 590 °C, ma wysoką pojemność cieplną 1,55 kJ/(kg K), gęstość 1,79 g/cm³ i jest tak rzadka jak woda (lepkość:2,1 mPa s). Bardzo łatwo zwilża powierzchnie metalowe, co może prowadzić do problemów z uszczelnieniem, jeśli konstrukcja i dobór materiałów jest nieodpowiedni. Stale nierdzewne są w dużej mierze odporne na roztopione azotany (szybkość erozji:6 - 15 µm / rok przy 570°C). Współczynnik przenikania ciepła na rurze przepływu turbulentnego wynosi około 6000 W/K m². Ze względu na wysoką pojemność cieplną (2,8 MJ/(Km3)), roztopiona saletra nadaje się również jako nośnik ciepła. Temperaturę topnienia można dodatkowo obniżyć przez dodanie azotynu sodu. Mieszanka soli 53% KNO3 o nazwie HiTec + 40% NaNO2 + 7% NaNO3 topi się w 140°C i ma szczególnie korzystne właściwości jako nośnik ciepła, jeśli toksyczność azotynu sodu jest nieistotna.
- Wanny saletrzane są stosowane do obróbki cieplnej przerabianych plastycznie stopów aluminium z zawartością magnezu do 10%. Maksymalna dopuszczalna temperatura stopionej soli zależy od zawartości magnezu; spada z 550°C przy 0,5% Mg do 380°C przy 10% Mg.
- w granatach dymnych
- w nawozie
Historia
Już w XI wieku saletra jest wymieniona w księdze Marcusa Graecusa, w której po raz pierwszy wspomniano również o mieszance czarnego prochu, jako nowej substancji zeskrobywanej z ziemi i kamieni. Książka Hassana ar-Rammaha z końca XIII wieku o walce konnej i użyciu machin wojennych (Al-Furusiyya wa al-Manasib al-Harbiyya ) zawiera już kilka przepisów dotyczących czyszczenia saletry popiołem drzewnym oraz przygotowania urządzeń zapalających i paliwa do rakiet.
Saletra była początkowo importowana z Indii; Wenecja czerpała duże zyski z handlu pośredniego. Wraz z rosnącym popytem i ze względu na niezależność, od końca XIV wieku rządy promowały własną produkcję saletry i zabezpieczały wszelkie prawa do produkcji, importu i użytkowania za pomocą drakońskich praw poprzez „reżim saletry”. Ze względu na szybkie uwalnianie tlenu saletra była podstawą nagłego spalania siarki i węgla drzewnego w prochu strzelniczym, a zatem, jako chronicznie deficytowa substancja, przez sześć wieków stanowiła strategiczny surowiec.
W Turyngii w XVI wieku działało dziewięć saletrni. Brzegi Wełtawy pod Pragą pokryto „ławkami sanitarnymi”, a miasto Halle udzieliło koncesji na wydobywanie saletry ze śmietników. Rosnący popyt na saletrę został częściowo zaspokojony przez dalszy import, głównie z Indii, oraz przez własne zakłady.
Od końca XIV wieku ogrody saletrzane były systematycznie uprawiane. Odchody zwierzęce (łajno, kał, mocz i krew) wypełniano ziemią wapienną, ziemią z cmentarzy, rzeźni lub bagien oraz wapnem, gruzem i popiołem w dołach lub spiętrzano i okazjonalnie zasypywano gnojowicą lub moczem. Po roku lub dwóch w wyniku rozkładu powstało tyle saletry, że można ją było wypłukać z ziemi. Wydajność wynosiła około 6:1, tj. 1 kg saletry uzyskano z 6 kg ziemi azotanowej.
Kotłownie saletrzane, jako zawód szczególny i bardzo niepopularny, mogły w każdej chwili wejść do posiadłości i tam szukać saletry. Dotyczyło to nawet kościołów w XVII i XVIII wieku, z wyłączeniem czasów nabożeństw. W Szwecji rolnicy musieli nawet płacić część podatków saletrą.
W XIX wieku doszło nawet do wojny azotanowej, którą Chile prowadziło z sąsiednimi krajami, aby zdobyć wyłączne posiadanie ogromnych pustynnych złóż azotanu sodu („Caliche”), które teraz można było natychmiast przekształcić za pomocą soli potasowych do postaci azotan potażu. Ten proces konwersji został ostatecznie zastąpiony od 1916 r. przez proces Habera-Boscha, polegający na wytwarzaniu amoniaku z powietrza i wody, a następnie konwersji do kwasu azotowego.
Źródła
- ↑ Wpis dotyczący azotanu potasu w bazie danych substancji GESTIS BGIA, pobranej 3 września 2007 r. (wymagany JavaScript)
Referencje
- Perry, R.H., Chemical Engineers' Handbook, wyd. 4, McGraw-Hill Book Company, Nowy Jork, 1963, s. 9-77
- Janz, GH i in., Fizyczne zestawienia danych dotyczących magazynowania energii II. Stopione sole, NSRDS, kwiecień 1979
- Gartz, J, kulturowa historia materiałów wybuchowych, E.S. Mittler &Son, Hamburg, 2006